Ewing Sarcoma in Adults
Ewing sarcoma is a type of bone or soft tissue cancer that primarily occurs in children and young adults. Often found in the long bones in the body, symptoms include pain, swelling and fever.
What is Ewing sarcoma?
Ewing sarcoma is cancer that occurs primarily in the bone or soft tissue. While Ewing sarcoma can develop in any bone, it is most often found in the hip bones, ribs, or long bones (e.g., femur (thighbone), tibia (shinbone) or humerus (upper arm bone)). It can involve the muscle and the soft tissues around the tumor as well. Ewing sarcoma cells can also metastasize (spread) to other areas of the body, including the bone marrow, lungs, kidneys, heart, adrenal glands and other soft tissues.
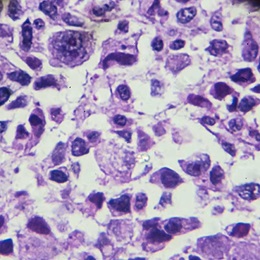
Under the microscope, Ewing sarcoma cells appear small, round and blue.
Ewing sarcoma gets its name from Dr. James Ewing, the doctor who first described the tumor in the 1920s.
Who gets Ewing Sarcoma?
As the second-most common type of bone cancer affecting children and young adults, it accounts for about 1 percent of childhood cancers. About 225 children and adolescents are diagnosed with Ewing sarcoma in the U.S. each year.
While Ewing sarcoma can occur at any time during childhood, it most commonly develops during puberty, when bones are growing rapidly. Ewing sarcoma most often occurs in children between the ages of 10 and 20. More males are affected than females. This type of cancer is uncommon in African-American, African and Chinese children.
Ewing sarcoma is a very rare cancer in adults.
What causes Ewing sarcoma?
The exact cause of Ewing sarcoma is not fully understood. Researchers have not been able to pinpoint risk factors or prevention measures for Ewing sarcoma. However, researchers have discovered chromosomal changes in a cell's DNA that can lead to the formation of Ewing sarcoma. These changes are not inherited. They develop in children for no apparent reason after they are born.
In most cases, the changes involve the fusing of genetic material between chromosomes 11 and 22. When a certain piece of chromosome 11 is placed next to the EWS gene on chromosome 22, the EWS gene gets "switched on." This activation leads to an overgrowth of the cells and eventually the development of cancer. Less often, there is an exchange of DNA between chromosome 22 and another chromosome that leads to the EWS gene being turned on. The exact mechanism remains unclear, but this important discovery has led to improvements in diagnosing Ewing sarcoma.
What are the symptoms of Ewing sarcoma?
Patients with Ewing sarcoma may experience symptoms differently. The most common symptoms include the following:
- Pain around the site of the tumor
- Swelling and/or redness around the site of the tumor
- Fever
- Weight loss and decreased appetite
- Fatigue
- Paralysis and/or incontinence (if the tumor is in the spinal region)
- Symptoms related to nerve compression from the tumor (e.g., numbness, tingling or paralysis)
The symptoms of Ewing sarcoma may resemble other medical conditions or problems. Always consult your doctor for a diagnosis.
How is Ewing sarcoma diagnosed?
In addition to a complete medical history and physical examination, diagnostic tests help confirm the presence of a tumor and also provide details about the tumor that can help oncologists determine the best approach to treatment. Diagnostic tests for Ewing sarcoma may include the following:
- Multiple imaging tests:
- X-ray. This diagnostic test uses invisible electromagnetic energy beams to produce images of internal tissues, bones and organs on film.
- Radionuclide bone scan. To begin, a small amount of radioactive dye is injected into the patient’s body and absorbed by the bone tissue. Pictures of the bone are captured with a special camera and are used to detect tumors and bone abnormalities.
- Magnetic resonance imaging (MRI). This diagnostic procedure uses a combination of large magnets, radio frequencies and a computer to produce detailed images of organs and structures within the body. This test is often done to see if the tumor has spread to the nearby soft tissues.
- Computed tomography (CT or CAT) scan. This diagnostic imaging procedure uses a combination of X-rays and computer technology to produce horizontal, or axial, images (often called slices) of the body. A CT scan shows detailed images of any part of the body, including the bones, muscles, fat and organs. While CT scans are more detailed than general X-rays, they are usually less detailed than MRI scans.
- Positron emission tomography (PET) scan. For this test, radioactive-tagged glucose (sugar) is injected into the bloodstream. Tissues that use the glucose more than normal tissues (such as tumors) can be detected by a scanning machine. PET scans can be used to find small tumors or to check if treatment for a known tumor is working.
- Blood tests, including blood chemistries
- Biopsy of the tumor. During this procedure, tissue samples are removed (with a needle or during surgery) from the body and examined under a microscope to determine if cancer or other abnormal cells are present.
- Bone marrow aspiration/biopsy. This procedure involves removing a small amount of bone marrow fluid and tissue, usually from part of the hip bone, to see if cancer has spread to the bone marrow.
Ewing sarcoma is difficult to distinguish from other similar tumors. Diagnosis is often made by excluding all other common solid tumors and using genetic studies.
What is the treatment for Ewing sarcoma?
The cancer care team develops specific treatment plans based on the following:
- The patient’s age, overall health and medical history
- The location and extent of the disease
- The patient’s tolerance to specific medications, procedures or therapies
- The expectations for the course of the disease
- The patient and/or parent’s opinion or preference
Depending on the stage of the tumor, treatment may include the following:
- Surgery to remove the tumor
- Chemotherapy
- Radiation therapy
- Amputation of the affected arm or leg
- Prosthetic fitting and training (if amputation occurs)
- Resections for metastases (e.g., pulmonary resections of cancer cells in the lung)
- Rehabilitation (e.g., physical and occupational therapy and psychosocial adaptation)
- Supportive care for the side effects of treatment
- Antibiotics to prevent and treat infections
- Continual follow-up care to determine the patient’s response to treatment, detect recurrent disease and manage late effects of treatment
Since Ewing sarcoma is rare in adults, the treatment details described in the following sections typically apply to children. Treating Ewing sarcoma in adults may involve modifications, particularly with chemotherapy, as children are much more tolerant of chemotherapy drugs.
Primitive neuroectodermal tumors (PNET) are very rare, molecularly-related tumors that often arise outside of the bone and are treated the same as Ewing sarcoma.
Before Ewing Sarcoma Treatment
Before treatment can begin, most patients will need to have their tumor staged. Typically, staging an Ewing tumor involves the following:
- A CT scan of the chest to rule out lung involvement
- An MRI of the affected area
- A bone or PET scan
- A bone marrow biopsy
All Ewing Sarcoma Treatment Begins with Chemotherapy
After the cancer stage is determined, patient care is taken over by a pediatric oncology team that will administer chemotherapy. All Ewing sarcoma patients require chemotherapy as the initial phase of therapy to shrink the primary or main tumor. Even if other aspects of treatment differ—like how the tumor is removed or treated locally—chemotherapy is always the first step. This is because doctors approach Ewing sarcoma as both a local and systemic disease. Chemotherapy is used to treat any potential metastasis (spread) to the lungs, which is quite common but very treatable. A multimodality approach is used even when the disease only appears to be localized at diagnosis.
The first set of chemotherapy drugs for Ewing sarcoma often includes vincristine, doxorubicin (Adriamycin) and cyclophosphamide (VAC). Following recovery from the first set of drugs, ifosfamide and etoposide (IE) may be given.
Like most other childhood cancers, the Children’s Oncology Group determines treatment protocols for Ewing sarcoma. Most cancer centers follow established protocols.
Surgery or Radiation to Treat the Tumor
Following initial chemotherapy to shrink the tumor, patients receive another MRI and CT scan of the chest to restage the tumor.
If the tumor is operable, the patient will usually have a resection (surgery). Generally, if cancer can be removed, surgery is recommended as an alternative to radiation, which can cause profound side effects, especially in young children.
If the tumor is inoperable, radiation may be required. Surgery may be discouraged in the following scenarios:
- The tumor is in a location where it’s unlikely that all tumor cells can be removed (e.g., the spine)
- The effects of surgery (e.g., living with paralysis or amputation) would significantly alter the patient’s quality of life
- There’s a high risk that function in certain body parts (e.g., the pelvis or wrist) cannot be restored
Sometimes both surgery and radiation are required. After tumor resection, the pathologist will analyze the tumor and look for a negative margin on the resected tissue. A negative margin indicates that the portion of tissue around the tumor does not have any live cancer cells. If any live cells are found, radiation is required as a follow-up treatment. Ewing tumors are typically very responsive to radiation.
Additional Chemotherapy
Another round of chemotherapy is given following surgery or radiation therapy to destroy tumor cells that may have spread to other parts of the body.
Targeted Therapies
Researchers are working to find new and improved ways to treat all kinds of cancers, including Ewing sarcoma. While some progress has been made, targeted treatments such as immunotherapy are considered experimental for patients with Ewing sarcoma. Doctors may pursue potential alternatives for patients with recurrent or advanced Ewing sarcoma who have already exhausted traditional treatment options.
Genomic sequencing may be used to find a drug that’s already FDA-approved for tumors with certain biomarkers (characteristics that may indicate that a tumor is a good target for a certain kind of therapy). Even drugs that haven’t previously been used to treat Ewing sarcoma may be considered.
Clinical trials may also be available. Your doctor can provide information about open studies at your cancer center or other centers across the country.
What kind of follow-up care is required after Ewing sarcoma treatment?
Ewing sarcoma patients will be monitored with X-rays of the original tumor every three to six months for three to five years. Similarly, patients will have regular CT scans of the lungs and periodic bone scans to detect recurrence as early as possible.
For the rest of their lives, patients will have yearly X-rays of the area of the original tumor to monitor any reconstructive devices and healing of the limb. Exercises may be suggested to increase the function of the affected limb.
What is the long-term prognosis for patients with Ewing sarcoma?
According to the American Cancer Society, the overall five-year survival rate for localized Ewing sarcoma is 70 percent. Patients with metastatic disease have a five-year survival rate of 15 percent to 30 percent.
It is unclear whether adults with Ewing sarcoma do as well as children with the condition. Some studies have suggested they do not. However, these studies have been criticized because they used lower doses of chemotherapy than those used in children. Other studies have suggested that when treated aggressively, adults can do just as well as children. The challenge with aggressive treatment is that adult bodies cannot withstand the same dosages of chemotherapy drugs that children can.
Factors that May Impact Prognosis
As with any cancer, prognosis and long-term survival can vary greatly from person to person. Since every individual is unique, your treatment and prognosis will be based on your unique health condition and needs. The individual patient prognosis for Ewing sarcoma greatly depends on the following:
- The extent of the disease
- The size and location of the tumor
- The presence or absence of metastasis
- The tumor's response to therapy
- The patient’s age and overall health (children often respond better to treatment than adults)
- The patient’s tolerance to specific medications, procedures or therapies
- New developments in treatment
Compared with smaller tumors, larger tumors are more difficult to remove and have had more opportunity to develop into micrometastatic disease. Micrometastasis describes cancer that has spread to other parts of the body as tumors that are too small to be detected.
Prompt medical attention and aggressive therapy help ensure the best possible prognosis. Continuous follow-up care is also essential.
Late Effects of Treatment
A person who was treated for Ewing sarcoma as a child or adolescent may develop health effects, which are called late effects, months or years after treatment ends. The type of late effects a survivor develops depends on the location of the tumor and the treatment method.
Late effects can include heart and lung problems, emotional and learning difficulties, growth issues and second malignancies associated with chemotherapy or radiation. For example, children treated for Ewing sarcoma have a higher risk than the average population of developing solid tumors or leukemia later in life.
Some treatments may later affect fertility. If this side effect is permanent, it will cause infertility (the inability to have children). Both men and women can be affected by fertility issues.
Ewing Sarcoma Recurrence
If Ewing sarcoma recurs, it usually happens within a few years of treatment. About 30 percent of patients will have a recurrence within the first five years.
Once cancer recurs, it becomes much more difficult to treat. The same chemotherapy drugs that were used during initial treatment cannot be used again due to toxicity concerns. Typically, a relapse of Ewing sarcoma prompts physicians to explore nontraditional or newer treatment options. As advances in targeted and immunotherapies continue, the hope is that prognosis for recurrent Ewing sarcoma will improve.
The Johns Hopkins Proton Therapy Center
Proton therapy is used to treat certain tumors in children and adults. Our treatment center, located at Sibley Memorial Hospital in Washington, D.C., combines advanced proton therapy technology, the latest research and caring specialists.