Polycystic Kidney Disease
What is polycystic kidney disease?
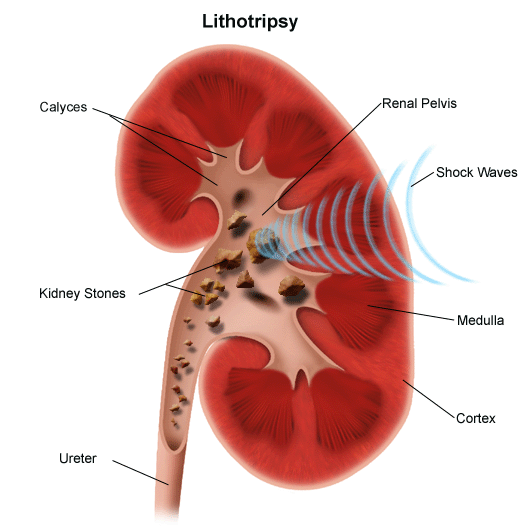
Polycystic kidney disease (PKD) is a genetic condition marked by the growth of numerous cysts (fluid-filled sacs) in the kidneys. The cysts become larger and the kidneys enlarge along with them. Slowly, the kidneys lose their ability to filter waste from the blood, which leads to progressive loss of kidney function and eventually to kidney failure. Some 600,000 people in the United States have PKD. PKD can also cause cysts in other organs, such as the liver.
There are two inherited forms of PKD:
-
Autosomal dominant (or adult) PKD is the most common form. Symptoms usually develop between the ages of 30 and 40, but they can begin during childhood. In this form of the disease, if one of the parents carries the disease gene, the child has a 50/50 chance of inheriting the disease.
-
Autosomal recessive PKD is a rare form. Symptoms begin in childhood and even in utero (before birth).
Symptoms of Autosomal Dominant PKD
In the early stages of the disease, people have few — if any — symptoms and relatively normal blood and urine tests, so the disease may be missed until it has progressed. Possible symptoms include:
-
Back pain and pain in the sides
-
Blood in the urine (hematuria)
-
Liver and/or pancreatic cysts
-
Heart valve abnormalities
Diagnosis
Autosomal dominant PKD is usually diagnosed by ultrasound of the kidneys, CT scans and MRI tests. The number and size of the cysts increase with age. Thus, even only two cysts in each kidney of a 30-year-old patient who also has a family history of the disease is a strong indicator. A genetic test to detect mutations is usually confirmatory but is not always necessary once symptoms develop.
Treatment
There is no cure for autosomal dominant PKD. Treatment involves managing symptoms (pain, headaches, high blood pressure, urinary tract infections) and preventing complications, as well as slowing the progression of the disease. End-stage kidney disease and kidney failure require dialysis and transplantation.
Symptoms of Autosomal Recessive PKD
A child with this form of the disease exhibits symptoms very early in life, even before birth. A child has a 25 percent risk of developing this disease if both parents carry the disease gene. A child must inherit two defective copies of the gene. Often, children with this disorder go on to develop kidney failure before reaching adulthood. In the most severe cases, newborns can die hours after birth because of respiratory failure. In milder cases, symptoms develop later in childhood and in early adulthood. Liver scarring is common in these cases.
Symptoms include:
-
High blood pressure
-
Urinary tract infections
-
Liver scarring
-
Low blood cell counts
-
Varicose veins
-
Stunted growth
Diagnosis
Ultrasound of the fetus can show enlarged kidneys. Imaging of the liver is also recommended as the disease scars the liver.
Treatment
There is no cure for this disease. Treatment focuses on preventing complications and alleviating symptoms. Medications are used to control high blood pressure and to treat urinary tract infections. Growth hormone can be used to improve growth. Dialysis and transplantation are needed once kidney failure develops.
When to call for help
If you suspect your child has PKD and you have family members with PKD, talk to your doctor.